
Thierry Briere :
www.chimie-briere.com
CINETIQUE CHIMIQUE
Soit la réaction :
a A + b B
à d D + e EExpérimentalement on peut suivre l’évolution des concentrations des réactifs (A et B) ou des produits (D et E) en fonction du temps. On obtient alors des courbes ayant l’allure suivante :
On définit la vitesse de la réaction comme la dérivée (ou la tangente) de ces courbes. Pour que les vitesses d’apparition (ou de disparition) soient les mêmes on divise par le coefficient stoèchiomètrique.
La vitesse de la réaction est donc définie par : v = (1/
ni) dCi / dt avec :n
i = coefficient stœchiométrique (positif pour les produits et négatif pour les réactifs).Ci = Molarité du composé i = [i]
Ici v = (-1/a) d[A] / dt = (- 1/b) d[B] / dt = (1/d) d[D] / dt = (1/e) d[E] / dt
Ordre d’une réaction – Equation de vitesse :
Expérimentalement on trouve que pour la plupart des réactions la vitesse peut se mettre sous la forme :
v = k [A]
a [B]b cette écriture est appelée équation de vitesse de la réaction.avec
k = constante de vitesse
a
= ordre partiel par rapport à Ab
= ordre partiel par rapport à Ba
+ b = ordre global de la réaction.Cas des réactions à un seul réactif
: Exemple : A à B + 2 CRéaction d’ordre 1 :
Définition de la vitesse : v = -d[A]/dt = d[B]/dt = ½ dC/dt
Equation de vitesse : v = k [A]
a = k [A] (par hypothèse a = 1)-d[A]/dt = k [A]
d[A] / [A] = - k dt
qui s’intègre en :
ln [A] = - k t + Cte
à t = 0 : [A] = [A]0
à Cte = [A]0ln [A] = - k t + ln [A]0
Si on porte ln [A] en fonction du temps on obtient une droite d’ordonnée à l’origine ln [A]0 et de pente -k.
ln [A] - ln [A]0 = - k t
ln ( [A] / [A]0 ) = - k t
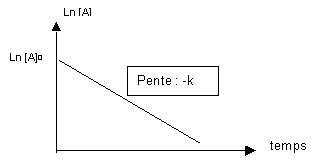
Si on porte ln ([A] / [A]0) en fonction du temps on obtient une droite passant par l’origine et de pente -k.
On peut aussi écrire sous forme exponentielle : [A] = [A]0 exp(-kt)
Unité de la constante de vitesse k : le logarithme n’ayant pas d’unité, k est l’inverse d’un temps.
Selon l’unité de temps utilisée, on aura k en s-1, min-1, h-1, j-1 etc.
Temps de demi-vie :
C’est le temps pour lequel la concentration du réactif à été divisée par deux.
[A] = [A]0 / 2 soit t1/2 = ln 2 / k ou k = ln2 / t1/2
Pour une réaction d’ordre 1 t1/2 ne dépend pas de la concentration initiale [A]0
Ln 2 = 0,69
Réaction d’ordre 2 :
Définition de la vitesse : v = -d[A] / dt = d[B] /dt = ½ d[C] / dt
Equation de vitesse : v = k [A] a = k [A]2 (par hypothèse a = 2)
-d[A]/dt = k [A]2
d[A] / [A]2 = - k dt
qui s’intègre en :
- (1 / [A]) = - k t + Cte
à t = 0 : [A] = [A]0 à Cte = -(1 / [A]0)
- (1 / [A]) = - k t - (1/ [A]0)
(1 / [A]) = k t + (1/ [A]0)
Si on porte (1 / [A]) en fonction du temps on obtient une droite d’ordonnée à l’origine (1/ [A]0) et de pente k.
- (1/[A]) + (1/ [A]0) = - k t
(1/[A]) - (1/ [A]0) = k t
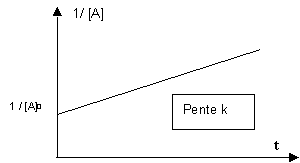
Si on porte (1/[A]) - (1/ [A]0)
en fonction du temps on obtient une droite passant par l’origine et de pente k.
Unité de la constante de vitesse k : k en t-1 (mol/L)-1 ou t-1 L Mol-1
Selon l’unité de temps utilisée : k en s-1 L mol-1, min-1 L mol-1, h-1 L mol-1 etc.
Temps de demi-vie :
C’est le temps pour lequel la concentration du réactif à été divisée par deux.
[A] = [A]0 / 2 soit 1/ [A] -1/ [A]0 = 1 / [A]0 et t1/2 = 1 / ( k [A]0 )
Pour une réaction d’ordre 2 t1/2 dépend de la concentration initiale [A]0
Cas des réactions à plusieurs réactifs
: Exemple : A + B à B + 2 CMéthode d’isolement d’Ostwald
:v = k [A]
a [B]bOn se ramène au cas précédants en utilisant un des réactifs en grand excès par rapport à l’autre.
Dans ces conditions la concentration du réactif en excès peut être considérée comme constante. Il y a dégénérescence de l’ordre.
A en excès [A]
a = cte v = k’ [B]b avec k’ = k [A]a0B en excès [B]
b = cte v = k’’ [A]a avec k’’ = k [B]b0On procède a deux expériences différentes dans lesquelles on détermine successivement
a et k’ puis b et k’’. On peut ensuite déduire l’ordre global et la constante de vitesse vraie k.Facteurs influençant la vitesse des réactions
:Les concentrations des réactifs influencent fortement la vitesse : plus les concentrations sont élevées et plus la vitesse est grande.
La température est aussi un facteur important, en général une augmentation de T augmente la vitesse de réaction.
Loi d’Arhenius : (Variation de k avec la température)
k = A exp (-Ea / RT) ou ln k = ln A – (Ea/RT)
A = facteur de fréquence de la réaction
Ea = Energie d’activation de la réaction
R = Cte des gaz parfait (8,31 j mol-1 K-1)
T = température absolue en Kelvins
Thierry Briere : www.chimie-briere.com