Thierry Briere : www.chimie-briere.com
P.C.E.M
RUDIMENTS DE THERMODYNAMIQUE CHIMIQUE
Aspect énergétique des réactions chimiques :
Au cours d'une réaction il peut y avoir : dégagement ou absorbtion de chaleur création d'énergie électrique etc
Une réaction qui dégage de la chaleur est dite exothermique.
Une réaction qui absorbe de la chaleur est dite endothermique.
Premier principe :
L'énergie ne peut être ni crée ni détruite. L'énergie ne peut que se transformer d'une sorte en une autre. Travail et Chaleur sont les formes les plus courantes de l'énergie.
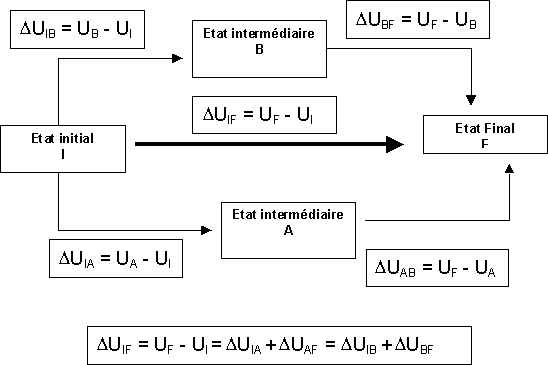
Fonction energie interne U :
Il existe une fonction appelée Energie Interne U. Cette fonction est une fonction d'état. La variation DU de cette fonction pour passer d'un état à l'autre ne dépend que de l'état initial et de l'état final du système et pas du chemin parcouru.
Cette fonction Energie Interne U est définie comme la somme Chaleur Q + Travail W
U = W + Q
DU = DW + DQ
dQ est la quantité de chaleur mise en jeu au cours de la transformation.
A Pression Constante (transformation isobare): dQP = Cp dT
A Volume Constant (transformation isochore): dQV = CV dT
Cp et Cv sont des coefficients appelés capacité calorifique molaire (en J mol-1) ou massique (en J g-1)
dW est la quantité de travail mise en jeu au cours de la transformation. dW = - P dV
dU = dQ - PdV
Unité de U : J mol-1
Fonction Enthalpie H
Cette fonction est aussi une fonction d'état . La variation DH de cette fonction pour passer d'un état à l'autre ne dépend que de l'état initial et de l'état final du système et pas du chemin parcouru.
Cette fonction Enthalpie est définie par :
H = U + PV
dH = dU + PdV + VdP
dH = dQ - PdV + PdV + VdP
dH = dQ + VdP
Si transformation isobare : P = Cte et dP = 0 soit dH = dQP = Cp dT
H correspond donc à la quantité de chaleur mise en jeu quand la réaction est effectuée à pression constante ce qui le plus souvent le cas. H = QP
Par convention :
* pour une réaction exothermique qui dégage de l'énergie, le DH correspondant est négatif.
* pour une réaction endothermique qui absorbe de l'énergie, le DH correspondant est positif.
Unité de H : J mol-1
Deuxième principe :
Fonction entropie S
On définit la fonction entropie S par dS = dQ / T
Cette fonction est aussi une fonction d'état. La variation DS de cette fonction pour passer d'un état à l'autre ne dépend que de l'état initial et de l'état final du système et pas du chemin parcouru.
A pression constante : dS = dQP / T = CP dT / T
L'entropie est une mesure du désordre du système : si le désordre augmente S augmente et DS > 0,si le désordre diminue S diminue et DS < 0.
Unité de S : J mol-1 K-1
Deuxième principe : L'entropie d'un système isolé qui n'échange rien avec l'extérieur ne peut que croître. L'entropie et le désordre de l'univers ne peuvent qu'augmenter. Pour mettre de l'ordre quel part on est toujours obligé de créer un plus grand désordre autre part.
Fonction Enthalpie Libre G :
On définit la fonction enthalpie libre ou énergie de Gibbs G par G = H - TS
Cette fonction est aussi une fonction d'état. La variation DG de cette fonction pour passer d'un état à l'autre ne dépend que de l'état initial et de l'état final du système et pas du chemin parcouru.
Cette fonction est utilisée pour prévoir comment va évoluer un système à pression constante.
La variation d'enthalpie libre DG d'une transformation spontanée est toujours négative.
Unité de G : J mol-1
Application aux réactions chimiques – Thermochimie
Soit la réaction chimique :
a A + b B = c C + d D
On peut définir un DRH un DRS et un DRG pour cette réaction.
Critères d'évolution (à P=Cte)
Si DRG < 0 : transformation thermodynamiquement favorisée = Réaction spontanée = Constante d'équilibre KR élevée ( KR > 1 )
Si DRG > 0 : transformation thermodynamiquement défavorisée = Réaction non spontanée = Constante d'équilibre KR faible ( KR < 1)
Si DRG = 0 : On est à l'état d'équilibre il n'y a pas d'évolution du système.
On se place très généralement dans les conditions standard à P = 1 bar, la température est très souvent prise égale à 298 K (25°C). Dans ces conditions, on définit des valeur standard de DH, DS et DG qu'on note avec un 0 en exposant à droite pour standard et un R en indice à droite pour réaction : DRG0298 , DRH0298 , DRS0298
Expression de DRG
On peut montrer que : DRG = DRG0 + RT ln QR
QR est le " monôme des activités " : QR = P aini
a A + b B = c C + d D
QR = ( aCc aDd ) / ( aAa aBb )
T = température absolue en Kelvins (K) : T( K ) = T(°C) + 273
R = Constante des gaz parfait = 8,31 J mol-1 K-1
L'expression de QR est identique à celle de KR déjà rencontrée si ce n'est que les activités utilisées sont les activités initiales au moment du mélange et non comme pour KR les activités à l'équilibre.
Il existe une relation directe entre KR et DG0R : DG0R = -R T ln(KR)
En effet
DRG = DRG0 + RT ln QR
A l'équilibre chimique on a : QR = KR et DRG = 0 soit 0 = DRG0 + RT ln KR
KR est une constante qui ne dépend que de la température.
Variations de H, S et G avec la température (à P = cte):
Variation de H :
dH = Cp dT
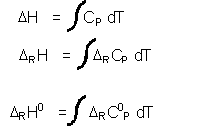
DRCP0 = S ni CPi0 = c CP0C + d CP0D - a CP0A - b CP0B
En général on fait l'approximation de considérer que les CPi0 sont indépendants de T.
On obtient alors après intégration :
DRH(T) = DRH(T0 )+ DRCP(T - T0)
DRH0(T) = DRH0(T0 )+ DRCP0 (T - T0) (Loi de Kirschoff)
En fait on fait très souvent l'approximation de négliger la variation de H avec T
Variation de S :
dS = CP dT / T
Par intégration en considérant les CP constants on obtient :
DRS(T) = DRS(T0) + DRCP ln ( T / T0)
En fait on fait très souvent l'approximation de négliger la variation de S avec T
Variation de G :
Si on fait l'approximation de négliger les variations de H et S avec T on obtient très facilement :
DRG(T) = DRH - T DRS
Variation de KR avec T : IMPORTANT
DRG0(T) = DRH0 - T DRS0 = -R T ln KR
ln KR = - DRH0 / R T + DRS0 / R
On suppose que DRH0 et DRS0 sont indépendants de la température
Si on porte ln KR en fonction de 1/T on obtient les valeurs de DRH0 et DRS0
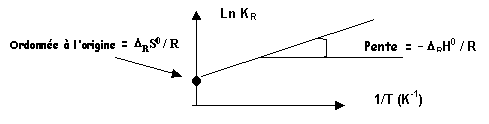
Il suffit donc de connaître la valeur de KR à deux températures pour connaître DRH0 et DRS0
La pente conduit à la valeur de DRH0
Pente = - DRH0 / R = D(lnK) / D(1/T) è DRH0 =- R D(lnK) / D(1/T)
L'ordonnée à l'origine conduit à la valeur de DRS0
Connaissant DRH0 on peut ensuite facilement calculer DRS0
soit graphiquement : O à O = DRS0 / R
On peut aussi calculer DRS0
DRG0 = -RT LnK = DRH0 – T DRS0
DRS0 = R Ln K + DRH0 / T
On peut écrire cette relation sous la forme différentielle (on dérive par rapport à T) :
[ d ( ln KR ) / dT ] = DRH0 / R T2 ( Loi de Vant'Hoff)
Cette relation permet de prévoir facilement l'influence de T sur un équilibre chimique.
RT2 est un terme obligatoirement positif.
Si DRH0 < 0 - Réaction exothermique : d lnKR et dT sont de signes contraires donc si T augmente KR diminue (et inversement).
Si DRH0 > 0 - Réaction endothermique : d lnKR et dT sont de même signes donc si T augmente KR augmente (et inversement).
Une élévation de température favorise donc le sens correspondant à la réaction endothermique.
Un abaissement de température favorise donc le sens correspondant à la réaction exothermique.
Tout se passe donc comme si l'équilibre cherchait à s'opposer à la variation de T c'est le principe de Le Chatelier.
Si une réaction possède un DRH0 nul, la température est sans influence sur l’équilibre.
Déplacement d'équilibre :
DRG = DRG0 + RT ln QR
DRG0 = - RT ln KR
DRG = - RT ln KR + RT ln QR
DRG = RT ln ( QR / KR )
Pour prévoir comment va évoluer un équilibre chimique sous l'effet d'un paramètre expérimental Variation de température ou modification du nombre de mole d'un des constituants il suffira de regarder comment varie QR au cours de la modification :
Si QR augmente DRG augmente et donc l'équilibre se déplace dans le sens 2.
Si QR diminue DRG diminue et donc l'équilibre se déplace dans le sens 1.
Si QR ne varie pas DRG ne varie pas et donc l'équilibre reste inchangé.
L'effet de T a été vu précédemment.
En général le principe de Le Chatelier est respecté c'est à dire que l'équilibre cherche à s'opposer aux variations. Ainsi, si on ajoute un corps l'équilibre va se déplacer dans le sens de disparition de ce corps. Inversement si on élimine un corps l'équilibre va se déplacer dans le sens de formation de ce corps.
Dans le cas où la réaction n’est pas effectuée a pression constante ce principe permet de prévoir l’effet d’une variation de pression sur l’équilibre : L’équilibre cherche à s’opposer à la variation de pression :
* si P augmente l’équilibre cherche à la faire diminuer en se déplaçant dans le sens de diminution du nombre de mole gazeuses.
* si P diminue l’équilibre cherche à la faire augmenter en se déplaçant dans le sens de l’augmentation du nombre de mole gazeuses.
* si au cours de la réaction le nombre de mole gazeuse ne varie pas, la pression est sans influence sur l’équilibre.
Calculs de grandeurs thermodynamiques :
Réaction de formation standard d'un corps pur :
C'est la réaction qui conduit à la formation d'une mole du corps pur dans son état standard sous P = 1 bar et à la température considérée à partir des corps simples qui le compose pris eux aussi, dans leur état standard sous P = 1 bar et à la température considérée. L'état standard correspondant à l'état le plus stable. (Dans 99 % des cas on se place à T = 298 K.)
Exemples :
Formation du méthane à 298 K : C (s) + 2 H2 (g) = CH4 (g)
Acide acétique à 298 K : 2 C (s) + 2 H2 (g) + O2(g) = CH3 COOH (l)
Acétate d'ammonium à 298 K : 2 C (s) + 7/2 H2 (g) + O2(g) + 1/2 N2(g) = CH3 COONH4 (s)
Pour ces réactions standards de formation on définit des enthalpies de formation standard DfH0, des entropies de formation standard Sf0, et des enthalpies libres de formation standard DfG0.
Ces grandeurs sont données dans des tables pour tous les corps.
On les utilise pour calculer facilement les grandeurs thermodynamiques associées aux réactions chimiques par la relation suivante :
DRX0 = S vi DfX0
aA + bB = cC + dD
DRH0 = S vi DfH0 = c DfH0 (C) + d DfH0 (D) - a DfH0 (A) - b DfH0 (B)
DRS0 = S vi S0 = c S0 (C) + d S0 (D) - a S0 (A) - b S0 (B)
DRG0 = S vi DfG0 = c DfG0 (C) + d DfG0 (D) - a DfG0 (A) - b DfG0 (B)
Remarque : pour les corps qui sont déjà dans leur état standard l'enthalpie de formation est par définition nulle : O2(g) - H2(g) - Fe(s) - Cl2(g) - S(s) - Br2(l) etc
Energies de liaison :
On définit l'énergie de dissociation d'une liaison chimique comme l'énergie qu'il faut fournir pour casser cette liaison. Cette grandeur est positive puisqu'il faut fournir de l'énergie.
Des tables donnent des valeurs moyennes de ces énergies de liaison. On utilise ces valeurs pour évaluer facilement les DRH0.
DRH0 = S El rompues - S El crées
Attention : pour pouvoir utiliser cette formule tous les corps doivent être gazeux.
Exemples :
CH4 (g) + O2(g) = CO2 (g) + H2O(l) DRH0298 = ?
1) utilisation des DfH0
DRH0298 = DfH0298CO2 (g) + DfH0298H2O(l) - DfH0298CH4 (g) - DfH0298O2 (g)
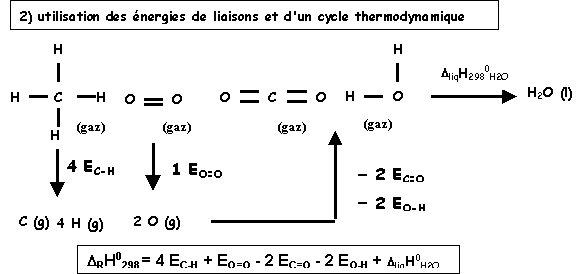
Combinaison linéaires de réactions chimiques
Quelques règles :
A + B = C + D : DRX0
C + D = A + B : - DRX0 (Renverser la réaction change le signe de DRX0)
2 A + 2 B = 2 C + 2 D : 2 DRX0 (Multiplier une réaction par un coefficient multiplie DRX0 par le même coefficient)
Exemple d'utilisation
R1 : A + 2 E = 2 F DRX01 = 100 KJ.mol-1
R2 : 1/2 F + 1/2 A = D DRX02 = - 50 KJ.mol-1
R3 : 3 F = B DRX03 = - 100 KJ.mol-1
R4 : 2 E + 2 D = B DRX04 = ??? KJ.mol-1
R4 = R1 - ( 2 * R2 ) + R3 DRX04 = DRX01 - 2 DRX02 + DRX03
DRX04 = 100 + 100 - 100 = 100 KJ mol-1
Vérification
A + 2 E + 2 D + 3 F = 2 F + F + A + B
2 E + 2 D = B
Cas des réactions d'oxydoréduction
On définit de manière un peu artificielle un DG0 pour les 1/2 réactions d'oxydoréductions.
Ox1 + n1 e- = Red1 : DG01 = - n1 F E01
Red2 = Ox2 + n2 e-: DG02 = + n2 F E02
Pour la réaction
DRG0 = n2 DG01 + n1 DG02
DRG0 = - n2n1 F E01+ n1n2 F E02 = n2n1 F ( E02 - E01 )
DRG0 = - RT ln KR = n2n1 F ( E02 - E01 )
RT ln KR = n2n1 F ( E01 - E02 )
2,3 RT log KR = n2n1 F DE0
(2,3 RT/ F) log KR = n2n1 DE0
0,06 log KR = n2n1 DE0
on retrouve bien sur la formule déjà rencontrée.
Autre intérêt : permet le calcul de valeurs de E0 de couples inconnus à partir du E0 de couples connus
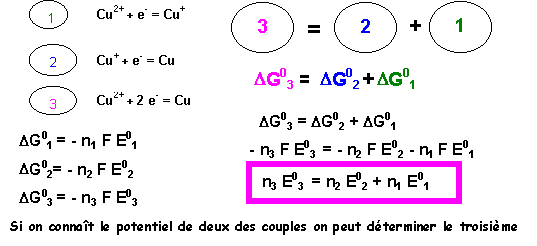
Thierry Briere : www.chimie-briere.com