Session 2001 : C.A. / P.L.P – CONCOURS EXTERNE -
MATHEMATIQUES –SCIENCES PHYSIQUES
CORRIGE DE L’EPREUVE DE CHIMIE (PARTIE A)
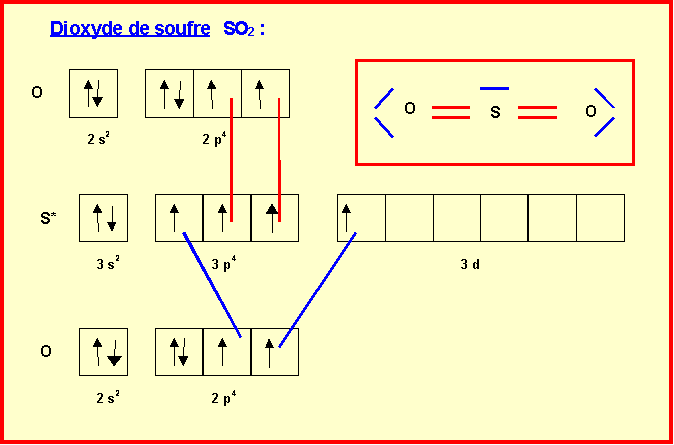
EXERCICE 1 – DIOXYDE DE SOUFRE
I-1 Structures
I-1 Structures électroniques
Oxygène (Z = 8)
O : 1s2 2s2 2p4
Soufre (Z = 16)
S : 1s2 2s2 2p6 3s2 3p4
I-2 : Eléments de la colonne 16
Oxygène (O) – Soufre (S)– Sélenium (Se) – Tellure (Te) – Polonium (Po)
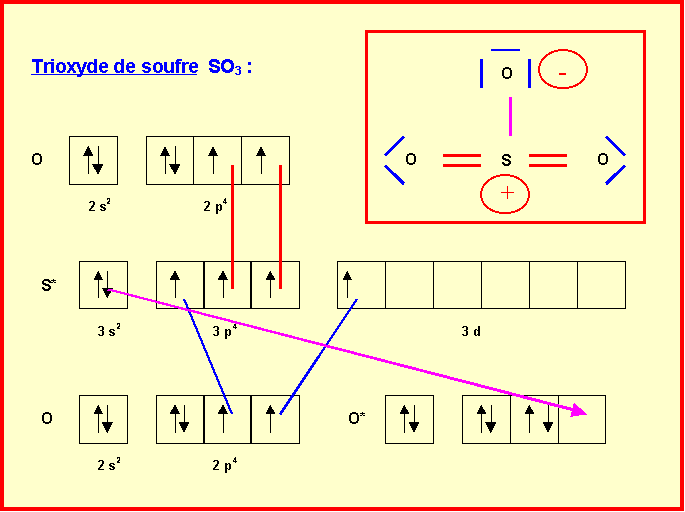
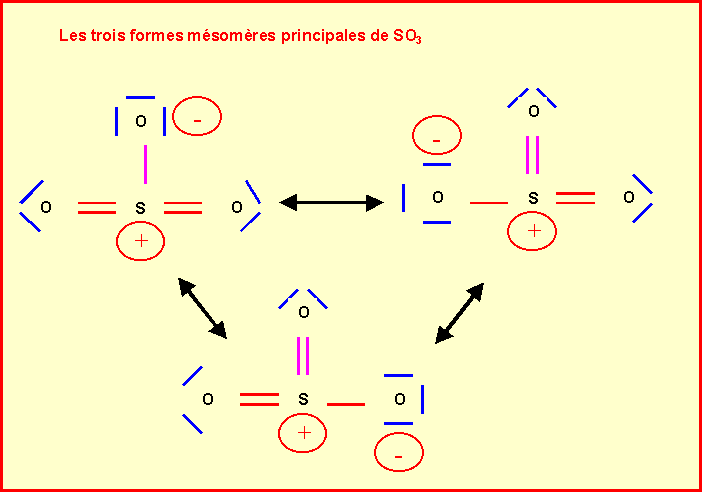
I-2 : Structures de Lewis de SO2 et SO3

II. Pollution et oxydes de soufre
II-1 : La principale source naturelle des oxydes de soufre est le volcanisme. La principale source artificielle est la combustion des combustibles fossiles (charbon, pétrole)
II-2 : Les oxydes de soufre réagissent avec l’eau pour donner des acides (H2SO3 et H2SO4). Le gaz SO2 présent dans l’atmosphère réagit donc avec l’eau atmosphérique, les acides formés (essentiellement H2SO3) retombent au sol avec la pluie ou la neige.
SO2+ H2O = H2SO3
Remarque : les principaux responsables des pluies acides sont plutôt les oxydes d’azote NO et NO2
III- Etude d’une étape de la synthèse de l’acide sulfurique
III-1 : Etude de la catalyse par l’oxyde de vanadium V2O5
II-1-a. Catalyseur : Substance qui modifie la vitesse d’une réaction chimique sans être transformée par la réaction. Le catalyseur modifie uniquement la vitesse (cinétique) de la réaction!; il n’agit pas sur l’équilibre thermodynamique : dans le cas d’une réaction thermodynamiquement possible, on obtient les mêmes produits et en mêmes proportions, avec ou sans catalyseur. À la fin de la réaction, on récupère intégralement le catalyseur. Un catalyseur est sélectif : il augmente la vitesse de certaines réactions seulement. Enfin, une substance qui catalyse une réaction chimique catalyse également sa réaction inverse.
III-1-b : Les réactifs étant gazeux et le catalyseur solide, il s’agit d’une catalyse hétérogène faisant intervenir deux phases différentes.
III-1-c. Un catalyseur est sans influence sur un équilibre chimique (voir question précédante)
III-2. Etude de l’équilibre 2 SO2(g) + O2(g) = 2 SO3(g)
2 SO2(g) + O2(g) = 2 SO3(g)
E.I 2 n0 n0 0
E.F 2 n0 - 2 x n0 - x 2 x
t = 2 x / 2 n0 = x / n0
x = t n0
nSO2 = 2 n0 - 2 x = 2 (n0 - t n0 ) = 2 n0 ( 1 – t )
nO2 = n0 - x = n0 - t n0 = n0 ( 1 – t )
nSO3 = 2 x = 2 n0 t
nt = nSO2 + nO2 + nSO3
nt = 2 n0 - 2 x + n0 - x + 2 x = 3 n0 – x = 3 n0 – t n0 = n0 ( 3 – t )
Fractions molaires :
XSO2 = nSO2 / nt = 2 n0 ( 1 – t ) / n0 ( 3 – t ) = 2 ( 1 – t ) / ( 3 – t )
XO2 = nO2 / nt = n0 ( 1 – t ) / n0 ( 3 – t ) = ( 1 – t ) / ( 3 – t )
XSO3 = nSO3 / nt = 2 n0 t / n0 ( 3 – t ) = 2 t / ( 3 – t )
Pression Partielles :
PSO2 = XSO2 Pt = 2 Pt ( 1 – t ) / ( 3 – t )
PO2 = XO2 Pt = Pt ( 1 – t ) / ( 3 – t )
PSO3 = XSO3 Pt = 2 Pt t / ( 3 – t )
Activités (avec Pt = P0 = 1 bar)
aSO2 = PSO2 / P0 = 2 Pt ( 1 – t ) / P0 ( 3 – t ) = 2 ( 1 – t ) / ( 3 – t )
aO2 = PO2 / P0 = Pt ( 1 – t ) / P0 ( 3 – t ) = ( 1 – t ) / ( 3 – t )
aSO3 = PSO3 / P0 = 2 Pt t / P0 ( 3 – t ) = 2 t / ( 3 – t )
Constante d’équilibre
KT = aSO32 aSO2-2 aO2-1
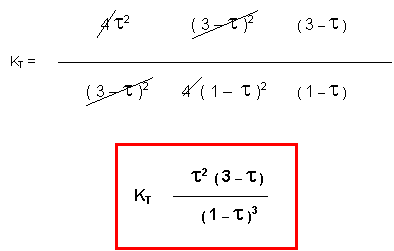
Enthalpie libre standard
DRG0T = - R T ln KT
Enthalpie et Entropie standard
DRG0T = DRH0T – T DRS0T
Si on suppose que DRH0 et DRS0 sont indépendant de la température et donc constants.
DRG0T = DRH0 – T DRS0
On peut calculer facilement DRH0 et DRS0 si on connaît la valeur de KT ou DRG0T pour deux températures différentes.
DRG0T1 = DRH0 – T1 DRS0
DRG0T2 = DRH0 – T2 DRS0
DRG0T2 - DRG0T1 = T1 DRS0 – T2 DRS0 = DRS0 [ T1 – T2 ]
DRS0 = [ DRG0T2 - DRG0T1 ] / [ T1 – T2 ]
DRH0 = DRG0T1 + T1 DRS0 ou DRH0 =DRG0T2 + T2 DRS0
Applications numériques
T (°C) |
T ( K ) |
t |
KT |
DRG0T (J mol-1) |
|
Réacteur 1 |
530 |
803 |
0,85 |
460,26 |
-40936,7 |
Réacteur 2 |
450 |
723 |
0,95 |
14801 |
-57720,5 |
DRS0 = - 209,8 J mol-1 K-1
DRH0 = - 209404,5 J mol-1 » - 209,4 kJ mol-1
Réponses aux questions :
III-2-a
PSO2 = XSO2 Pt = 2 Pt ( 1 – t ) / ( 3 – t )
PO2 = XO2 Pt = Pt ( 1 – t ) / ( 3 – t )
PSO3 = XSO3 Pt = 2 Pt t / ( 3 – t )
III-2-b
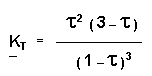
III-2-c
T = 530 °C = 803 K : K803 = 426,6
T = 450 °C = 723 K : K803 = 14801
III-2-d-1
DRH0 = - 209404,5 J mol-1 » - 209,4 kJ mol-1
Remarque : on aurait pu aussi déterminer DRH0 en utilisant la loi de Van’t Hoff
d lnK / dT = DRH0 / RT2
d lnK = { DRH0 / R } dT / T2
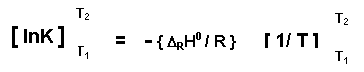
Ln ( KT2 / KT1 ) = - { DRH0 / R } [ 1/T2 – 1/T1 ]
DRH0 = - R Ln ( KT2 / KT1 ) / [ 1/T2 – 1/T1 ]
A.N : DRH0 = - 209,4 kJ mol-1 (on retrouve le même résultat)
III-2-d-2
DRH0 < 0 : la réaction est exothermique.
On peut prévoir l’influence d’une diminution de la Température soit par le principe de Le Chatelier soit par la loi de Van’t Hoff.
Principe de Le Chatelier : Si on diminue la température, l’équilibre se déplace dans le sens exothermique, soit ici dans le sens 1 de formation de SO3, le taux de conversion doit donc augmenter quand la température diminue.
C’est bien ce que l’on observe expérimentalement ( T2 < T1 et t2 > t1 )
Loi de Van’t Hoff :
d LnK / dT = DRH0 / RT2
R et T2 sont deux grandeurs positives
Ici DRH0 est négatif.
d ln K et dT sont donc de signes opposés.
Si T diminue, dT < 0 et donc d Ln K > 0
Donc Ln K et K augmentent quand T diminue. Cela revient à un déplacement de l’équilibre dans de formation de SO3 et donc à une augmentation de t.
III-2-e
Enthalpie libre standard
DRG0T = - R T ln KT
DRG0T1 = DRG0 (530) = -57720,5 J mol-1
DRG0T2 = DRG0 (450) = -40936,7 J mol-1
III-2-f
DRS0 = - 209,8 J mol-1 K-1
Remarques :
1) On aurait pu calculer DRS0 à partir des valeurs de DRG0T et DRH0.
DRG0T = DRH0 – T DRS0
DRS0 = { DRH0 – DRG0T } / T
Le calcul conduit au même résultat pour les deux températures.
2) DRS0 < 0 : Le signe négatif indique une diminution d’entropie at donc une augmentation de l’ordre au cours de la réaction. Cela est du à la diminution du nombre de mole gazeuses au cours de celle-ci.
IV - Le procédé Clauss : une valorisation des oxydes de soufre
SO2 peut oxyder H2S.
SO2 + 4 H+ + 4 e- = S + 2 H2O (E0 = 0,43 V)
2 * [ H2S = S + 2 H+ + 2 e- ] (E0 = 0,14 V)
SO2 + 4 H+ + 4 e- + 2 H2S = S + 2 H2O + 2 S + 4 H+ + 4 e-
SO2 + 2 H2S = 3 S + 2 H2O
iV-1
L’équation bilan est :
SO2 + 2 H2S = 3 S + 2 H2O
iV-2
Espèce |
S |
H2S |
SO2 |
N.O de S |
0 |
-II |
+ IV |
Cas de SO2
O est plus électronégatif que S et " prend " donc les doublets des liaisons O -S.
S " possède " donc 2 électrons dans la molécule SO2
Il en " possède " normalement 6 et il a donc " perdu " 4 électrons soit un degré d’oxydation de +4
Cas de H2S
S est plus électronégatif que H et " prend " donc les doublets des liaisons H -S.
S " possède " donc 8 électrons dans la molécule H2S
Il en " possède " normalement 6 et il a donc " gagné " 2 électrons soit un degré d’oxydation de –2
Cas de S
S étant seul ne " perd " ni ne " gagne " d’électrons et à donc un nombre d’oxydation nul.
IV-3 :
Il s’agit d’une réaction d’oxydoréduction.
Sa seule particularité est que les deux espèces réagissantes ont une même espèce conjuguée.
Le meilleur oxydant SO2 à pour réducteur conjugué S
Le meilleur réducteur H2S à pour oxydant conjugué la même espèce S .
Comme il s’agit de la réaction inverse d’une réaction de dismutation on lui donne parfois le nom de réaction de rétro-dismutation ou d’amphotérisation.
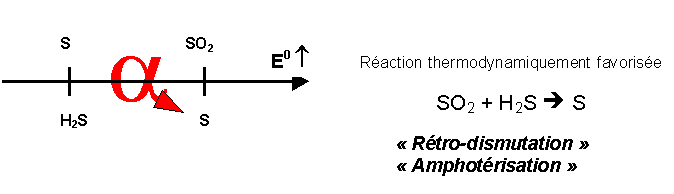
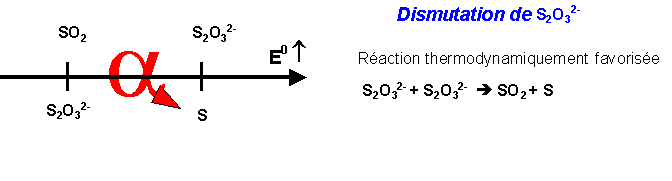
Rappel sur la dismutation :
Dans une réaction de dismutation une espèce A appartenant à deux couples différents réagit sur elle même.
L’espèce A est a la fois oxydante et réductrice (amphotère) et le potentiel du couple dans lequel elle est oxydante est supérieur à celui dans lequel elle est réductrice comme dans le schéma suivant.
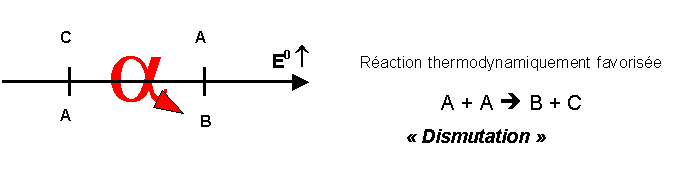
Au cours de la réaction l’espèce amphotère A est a la fois oxydée et réduite. On passe d’un degré d’oxydation intermédiaire a un degré plus élevé et un degré plus faible.
Dans notre exemple, le soufre S (N.O=0) est " intermédiaire " entre SO2 (N.O=+IV) et H2S (N.O=-II). La réaction étudiée est donc bien la réaction inverse de la dismutation du soufre.
V - Diagramme potentiel-pH du soufre
V-1 : Domaines de prédominances des espèces
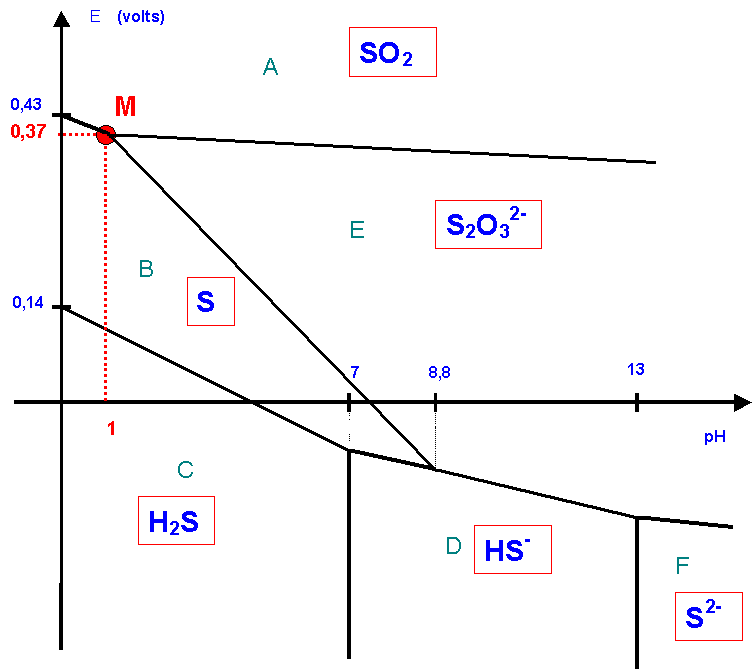
Pour placer correctement les diverses espèces, il faut savoir qu’elles se placent de la manière suivante :
Lecture horizontale à E constant
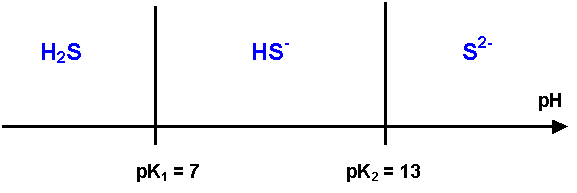
De gauche à droite on place les espèces de même degré d’oxydation de la plus acide à gauche à la plus basique à droite, dans notre cas H2S puis HS- puis S2-.
Cela se justifie par le diagramme de prédominance acidobasique.
Lecture verticale à pH constant
De bas en haut on place les divers nombre d’oxydation du plus faible en bas au plus élevé en haut.
Dans notre cas les divers nombre d’oxydation sont :
Espèce |
S |
H2S - HS- - S2- |
SO2 |
S2O32- |
N.O de S |
0 |
-II |
+ IV |
0 et +IV |
Cas de S2O32-

V-2 : Couples S2O32- / S et SO2 / S2O32-
V-2-a
On suppose que toutes les espèces sont en solution aqueuse à la concentration de 1 mol.L-1, sauf S qui est considéré comme solide et H3O+ dont la concentration est évidemment variable.
Couple S2O32- / S
S2O32- + 6 H+ + 4 e- = 2 S + 3 H2O
E = E0 + ( 0,06 / 4 ) log { [ S2O32- ] [H+]6 }
E = E0 + ( 0,06 / 4 ) log [ S2O32- ] + ( 0,06 / 4 ) log [H+]6
E = E0 + 0,06 log [ S2O32- ] + 0,09 log [H+]
E = E0 + 0,06 log [ S2O32- ] - 0,09 pH
E = 0,46 - 0,09 pH
Couple SO2 / S2O32-
2 SO2 + 2 H+ + 4 e- = S2O32- + H2O
E = E0 + ( 0,06 / 4 ) log { [ SO2 ] [H+]2 / [ S2O32- ] }
E = E0 + ( 0,06 / 4 ) log { [ SO2 ] / [ S2O32- ] } + ( 0,06 / 4 ) log [H+]2
E = E0 + 0,06 log [ S2O32- ] + 0,03 log [H+]
E = E0 + 0,06 log [ S2O32- ] - 0,03 pH
E = 0,40 - 0,03 pH
V-2-b : Intersection des droites
0,46 - 0,09 pH = 0,40 - 0,03 pH
0,46 - 0,40 = 0,09 pH - 0,03 pH
0,06 = 0,06 pH
pH = 1
E = 0,46 - 0,09 = 0,40 - 0,03 = 0,37 v
Les deux droites se coupent donc au point M de coordonnées :
pH = 1 et E = 0,37 v
V-2-c : Dismutation de S2O32-
Pour un pH < 1, le potentiel du couple SO2 / S2O32- est inférieur à celui du couple S2O32- / S.
Par exemple à pH = 0 : E SO2 / S2O32- = 0,40 v et E S2O32- / S = 0,46 v.
S2O32- peut donc réagir sur lui même et se dismuter. A pH très acide, l’ion S2O32- n’est donc pas stable en solution aqueuse.
S2O32- + 6 H+ + 4 e- = 2 S + 3 H2O
S2O32- + H2O = 2 SO2 + 2 H+ + 4 e-
S2O32- + 6 H+ + 4 e- + S2O32- + H2O = 2 S + 3 H2O + 2 SO2 + 2 H+ + 4 e-
2 S2O32- + 4 H+ = 2 S + 2 H2O + 2 SO2
EXERCICE II – ACIDE BENZOÏQUE
I- Structure de l’acide benzoïque
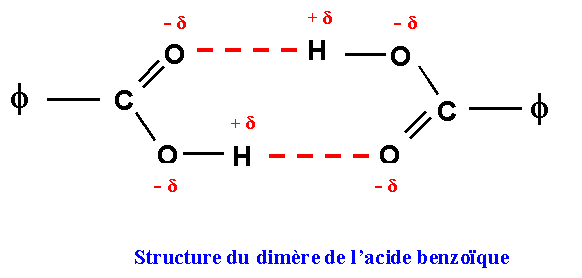
La masse molaire moyenne est deux fois plus élevée que la masse molaire vraie de f - COOH , cela indique l’existence d’un dimère. Celui-ci est formé par l’association de deux molécules de f - COOH par Liaison Hydrogène. La liaison Hydrogène est due à l’attraction électrostatique entre dipôles permanents (force de Keesom). Cette interaction se rencontre dans les molécules comportant des atomes d'hydrogène liés à des atomes très électronégatifs F, O et N essentiellement. On peut alors parler de véritables associations intermoléculaires qui peuvent même subsister à l'état gazeux. Dans ce cas les interactions sont si fortes, que l'ordre de grandeur de l'énergie associée atteint de 10 à 30 KJ mol-1 ce qui les rapprochent des liaisons de covalence (100 KJ mol-1) et justifie leur nom de Liaison Hydrogène.
L'existence de liaisons hydrogène va entraîner des modifications importantes des propriétés moléculaires. L'énergie des liaisons hydrogène étant beaucoup plus faible que celles des liaisons covalentes normales leur longueur sera sensiblement plus élevée de l'ordre de 2 à 3 Å. On représente généralement les liaisons Hydrogène par des traits pointillés.
II- Propriétés de l’acide benzoïque
II-1 pH d’une solution d’acide benzoïque
Appelons AH l’acide benzoïque.
Dans l’eau s’établit l’équilibre de dissociation de cet acide faible :
AH + H2O = A- + H3O+
La relation de base est celle reliant [H3O+] ; [OH-] ; [A-] et [AH] par l'intermédiaire de Ka.
Ka = [H3O+] [A-] / [AH]
Avec de plus :
Electroneutralité de la solution : [A-] = [H3O+] - [OH-]
Conservation de la matière : [AH] = C0 - [A-]
Voyons comment cette relation fondamentale peut se simplifier.
Approximation 1 :
On étudie le pH d'une solution acide et on peut donc considérer que dans une telle solution la concentration des ions Hydroxydes est inférieure à celle des ions Hydronium.
Si la concentration de l'acide n'est pas trop faible cette approximation sera valable et on pourra négliger [OH-] devant [H3O+]
[OH-] <<< [H3O+]
[A-] = [H3O+] - [OH-] si on néglige [OH-] devant [H3O+] on obtient : [A-] = [H3O+]
[AH] = C0 - [A-] = C0 - [H3O+]
Ka = [H3O+]2 / {C0 - [H3O+]} : Expression simplifiée valable si [OH-] <<< [H3O+]
Approximation 2
L'acide est faible et est donc par définition peu dissocié dans l'eau. Cela signifie qu'a priori il est présent en solution essentiellement sous forme acide AH et très peu sous forme basique A-. On pourra donc négliger [A-] devant [AH] et donc [AH] = C0 - [A-] se simplifiera en C0 = [AH].
Si les deux approximations sont simultanément vérifiées on peut donc simplifier énormément le problème.
Ka = [H3O+]2 / C0
Soit : [H3O+]2 = Ka C0
2 log [H3O+] = log Ka - log C0
-2 pH = log Ka - log C0
soit finalement : pH = 1/2 { pKa + pC }
Les conditions de validité de cette formule simplifiée sont d'une part que [OH-] soit négligeable devant [H3O+] et d'autre part que [A-] soit négligeable devant [AH].
La première condition est pratiquement toujours réalisée sauf pour des solutions extrêmement diluées de pC proche de 7. En revanche, la deuxième condition est plus exigeante, en effet on peut montrer facilement que pour que cette condition soit satisfaite il faut que pH < pKa - 1.
Ka = [A-] [H3O+] / [AH]
[H3O+] = Ka [AH]/[A-]
log [H3O+] = log Ka + log { [AH] / [A-] }
pH = pKa + log { [A-] / [AH] }
Si on considère que [A-] est négligeable devant [AH] si [A-] < 0,1 [AH]
alors { [A-] / [AH] } < 0,1 et log { [A-] / [AH] } < -1 soit pH < pKa - 1
Il est donc évident que la formule simplifiée ne "marchera" qu'en milieu relativement acide c'est a dire pour des concentrations C0 élevées et donc pC faibles.
Dans la pratique tout dépend du degré de précision attendu nous allons voir que la formule est correcte pour :
pC < pKa - 0,5 à 0,1 unité de pH près.
pC < pKa - 1 à 0,05 unité de pH près.
pC < pKa - 1,5 à 0,01 unité de pH près.
Dans notre cas pC = - log 0,5 = 0,301 est très faible et la solution sera suffisamment acide pour que l’approximation 2 soit valable.
A.N : pH = ½ ( 4,2 + 0,301) = 2,25 qui est bien nettement inférieur à pKa – 1 = 3,2.
Conclusion : pH = 2,25
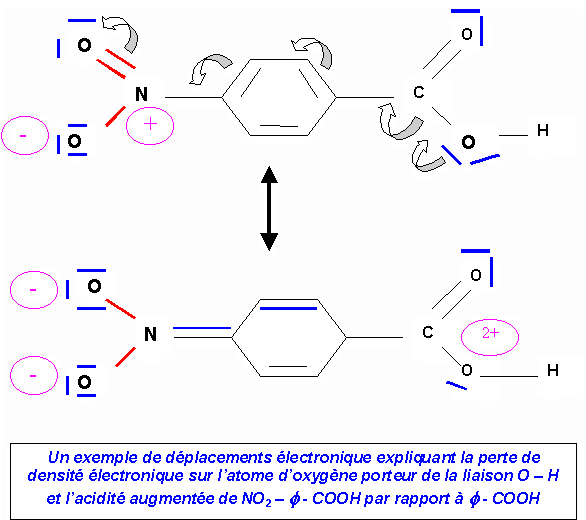
II-2 : Comparaison acide benzoïque / acide paranitrobenzoïque
Le pKa de l’acide paranitrobenzoïque (pKa = 3,4) est inférieur à celui de l’acide benzoïque (pKa = 4,2). L’acide paranitrobenzoïque est donc un acide plus fort que l’acide benzoïque. Cela s’explique par le fort effet mésomère attracteur du groupement nitro qui diminue la densité électronique au niveau de l’atome d’oxygène porteur de la liaison O – H ce qui facilite le départ d’un ion H+ et augmente donc l’acidité de
NO2 – f - COOH.
.
III- Synthèse de l’acide benzoïque
II-1-a : schéma du montage
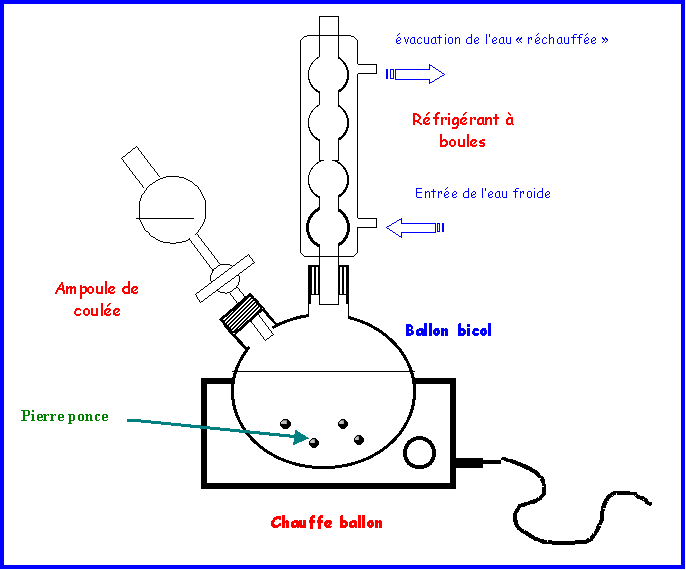
III-1-b : Rôle de la pierre ponce
La pierre Ponce, est une roche ignée ayant une texture spongieuse et mousseuse, composée principalement de verre. Elle est souvent constituée de fibres ou fils parallèles, avec des cavités formant une structure délicate, légère et très dure. La pierre ponce est produite par l'expansion des gaz de lave occlus ou internes quand ils atteignent la surface de la Terre. Elle se développe abondamment dans les laves de rhyolite, car celles-ci sont habituellement très visqueuses. On la trouve, par exemple, autour des volcans l'Etna, le Vésuve et le Stromboli, en Italie. On utilise parfois la pierre ponce comme abrasif. En chimie elle est utilisée lors de chauffage de liquides pour régulariser l’ébullition. De par sa structure spongieuse elle contient de l’air qui lors du chauffage est libéré sous de micro-bulles ; c’est cet apport régulier de petites bulles qui régularise l’ébullition. Au cours de l'ébullition, il se forme de la vapeur au sein du liquide. Lorsque ces bulles de vapeur remontent dans le liquide, elles créent une turbulence et le bouillonnement associé à l'ébullition. En l’absence de gaz dissous de grosses bulles de vapeur se forment au fond du récipient, là ou la température est la plus élevée. Ces grosses bulles " explosent " et provoquent des projections gênantes. En présence de gaz dissous ou de pierre ponce on évite une ébullition brutale et ces projections intempestives.
III-1-c : Rôle du chauffage
La vitesse des réactions chimique (cinétique chimique) dépend fortement de la température et très généralement augmente avec celle-ci, c’est donc pour augmenter la vitesse de réaction qu’on procède à température élevée.
III-1-d : Rôle du réfrigérant
Le réfrigérant sert à condenser les vapeurs qui se forment et donc à éviter leur départ dans l’atmosphère, source de pollution et de forte diminution du rendement si les produits ou les réactifs sont volatils. On utilise généralement un réfrigérant à boules en position verticale lors des chauffage à reflux, les boules permettent une grande surface de contact entre les vapeurs et les parois refroidies. Généralement on utilise l’eau froide comme fluide réfrigérant, l’arrivée de l’eau froide doit se faire par le bas du réfrigérant puisque c’est là que la température est la plus élevée. Les vapeurs condensées retombent dans le ballon, une position verticale évite que le liquide ne reste piégé dans les boules.
III-2 : Etude des espèces mises en jeu
III-2-a :
L’alcool benzylique f – CH2 OH est un alcool primaire aromatique.
III-2-b :
Il se forme un précipité marron de dioxyde de manganèse MnO2
III-2-c :
Le milieu est fortement basique puisque l’on à affaire à une solution de soude à environ 2 mol.L-1 (soit un pH de l’ordre de 14 !). L’acide benzoîque (pKa = 4,2) sera donc majoritairement sous forme de son sel de sodium dissous ( f -COO- Na+ ).
III-2-d :
On fait réagir f – CH2 OH et MnO4- en milieu basique.
Les couples en présence sont : f – CO O- / f – CH2 OH et MnO4- / MnO2
MnO4- va oxyder f – CH2 OH

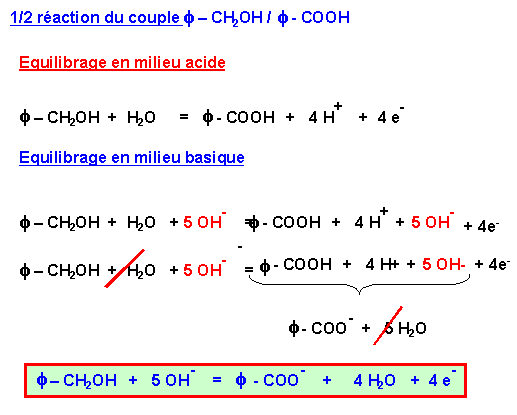
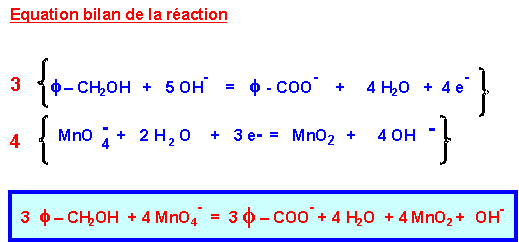
III-2-e Réactif en excès
Quantité de matière MnO4-
150 mL de solution à 0,3 mol.L-1
nMnO4- = 150 * 0,3 10-3 = 0,045 mol
Quantité de matière f - CH2OH
3 mL soit 3 * 1,04 = 3,12 g soit 3,12 / 108 = 0,0289 mol
Réactif en excés
D’après l’équation bilan : 3 moles de f - CH2OH réagissent avec 4 moles de MnO4-
0,0289 mole de f - CH2OH réagiront donc avec 4 * 0,0289 / 3 = 0,0385 mole de MnO4-
L’ion permanganate est donc en excès.
II-2-f
L’ajout d’éthanol permet l’élimination de l’excès de MnO4- selon la réaction :
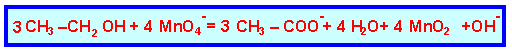
III – Obtention de l’acide benzoïque
III-3-a
L’intérêt principal de la filtration avec tirage sous vide selon la technique dite de büchner est sa rapidité bien plus grande.
II-3-b :
On ajoute de l’acide concentré afin de diminuer fortement le pH de la solution. Quand le pH deviendra inférieur au pKa de l’acide benzoïque (soit pH < 4,2), l’espèce prédominante sera la forme acide f - COOH.
En effet on a : pH = pKa + log { [f - COO- ] / [f - COOH ] }
Dans la pratique, si on se place par exemple à pH = pKa – 2 = 2,2 la concentration de la forme basique sera 1/100 de celle de la forme acide.
La forme acide électriquement neutre est beaucoup moins soluble que la forme chargée basique, on observe donc sa précipitation dans le milieu et il est alors facile de l’isoler par filtration.
III-3-c : Rendement de la synthèse
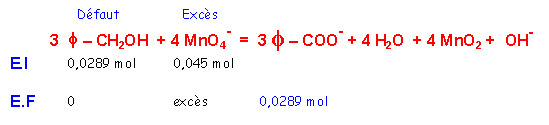
Si la réaction était totale et qu’il n’y ait aucune perte au cours des diverses opérations on devrait obtenir 0,0289 mole d’acide benzoïque soit 0,0289 * 122 » 3,53 g.
Le rendement serait alors de 100%.
On obtient en fait seulement 2,82 g d’acide benzoïque.
Le rendement de la synthèse est donc : 2,82 / 3,53 » 0,8 soit 80 %.
II-3-d Caractérisation de l’acide benzoïque
Le produit obtenu étant un solide la technique la plus simple et la plus couramment utilisée est la mesure du point de fusion et sa comparaison avec celui du produit pur donné dans les tables.
Il sera tout de même nécessaire de procéder au préalable a une purification par recristallisation dans l’eau chaude.
On pourrait aussi utiliser une technique spectroscopique : Résonance Magnétique nucléaire ou spectre Infra-Rouge par exemple.
IV- Synthèse d’un ester à partir de l’acide benzoïque et du méthanol
IV-1
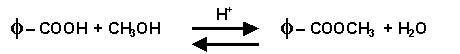
L’ester formé est le benzoate de méthyle.
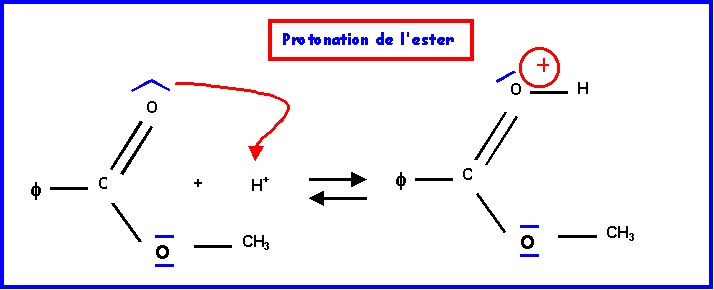
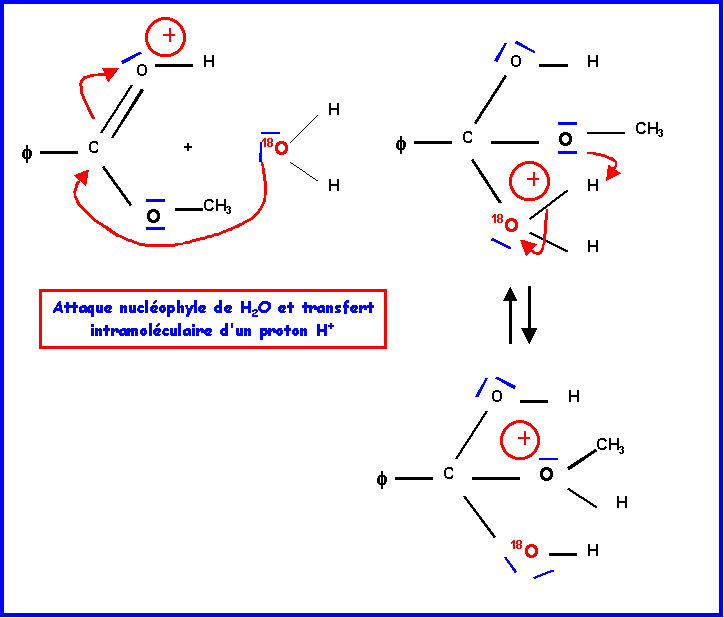
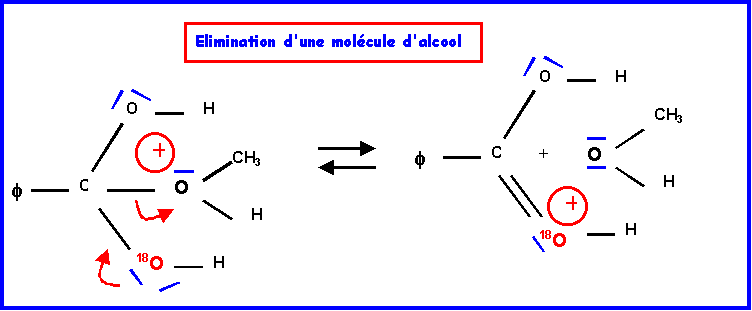
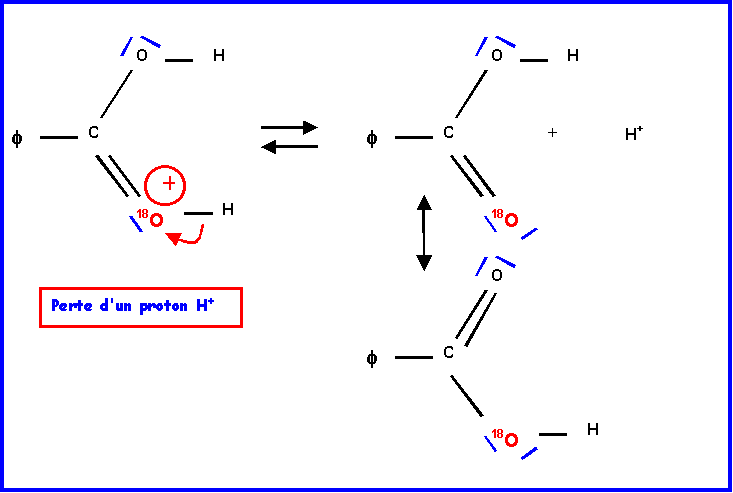
IV-2-a : Mécanisme réactionnel
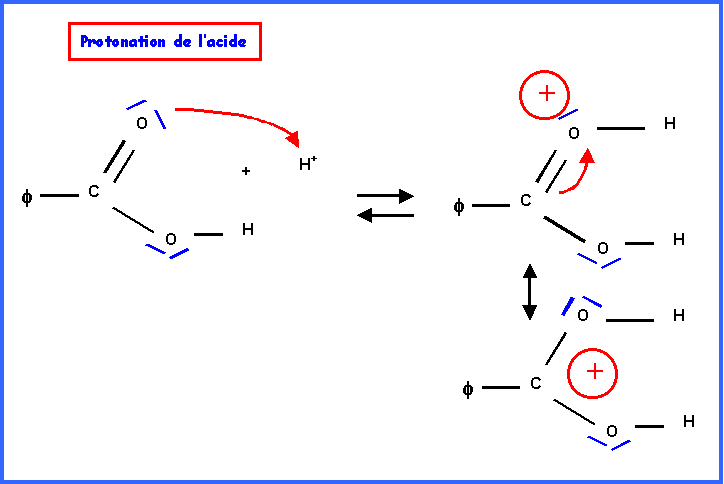
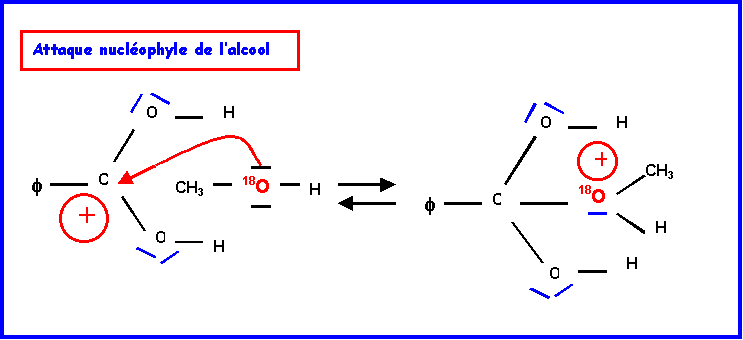
![]()
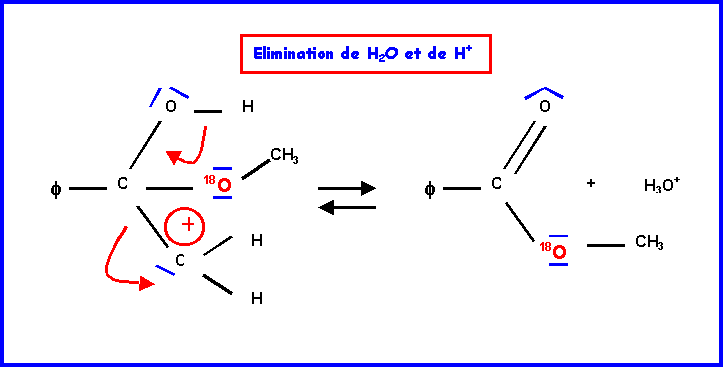
IV-2-b : Hydrolyse de l’ester ( par H218O )
L’oxygène marqué 18O se trouve dans la molécule d’acide.